
The Amazing Accessory Genome in S. pneumoniae
by Bill Hanage
It’s been remarked before, but bacteria are
different. Really different. I
can think of few more potent demonstrations of this than this new
paper
that compares accessory genomes in populations of pneumococci – my favorite
bug. It is also true that the results of it are so weird that even some of the
authors took a while to come to the ‘fall off your chair moment’ as one of them
eloquently put it. So I am writing this, in the hope that you will fall off
your chair a little earlier than you would have done otherwise. Also if you’re
not a bacterial population genomics fan already, I hope this convinces you to
become one.
This is work driven by Jukka Corander and Nick Croucher. Yes I am on the paper, but all the best bits of it are down to them, not me.
How much can strains vary in gene content, and why? Microbiologists reading this will likely know it already, but if not let me point you in the direction of Welch et al 2002 which compared three E coli and found that less than 40% of the genes were in all three. That still blows me away. Similar variation has been found in most bacteria where we’ve looked. And the pneumococcus is no exception; we and others have documented extensive variation in gene content.
What that gene content means is another thing. Initially I wanted to look at this by comparing divergence in the core genome, with divergence in the accessory. The idea is that we can calibrate how quickly we expect two lineages to become more dissimilar in gene content as they diverge. Such things can happen quite quickly (Look at this, in which Yonatan Grad asked how much change happened between very closely related E. coli). With the pneumococcus, we examined a very carefully sampled population from kids, here in the Commonwealth of Massachusetts. What we found was that closely related things had pretty similar accessory genomes, and more distantly related things had pretty different accessory genomes, with little inbetween.
This made me all excited, as I thought that these differences could be related to ecology. People think (rightly or wrongly) of the accessory genome as interesting and ecologically relevant. So this observation could be explained by saying that this is how different things had to be in terms of gene content, in order to be adapted to a unique niche. Nick squelched that idea (as he has so many of my ideas) with a fact: my simple notion posited that each lineage was characterized by accessory loci which constituted its own ‘ecological address’ in niche space so that they wouldn’t compete with other lineages. But in fact lineages had different combinations of the same accessory loci. Like they’d downloaded three each of the same 10 apps to their phones. Later we showed that the pattern could also be explained by recombination of accessory elements that had little consequence for fitness.
Could be. But not could only be.
What the new work does is compare the Massachusetts sample with other large collections from Thailand, the UK and the Netherlands. This is comparative population genomics, where we are interested in how similar or otherwise the populations are. One way to do that is ask whether the same lineages are everywhere, as you might expect in the case where there is lots of migration, and the selective pressures are similar everywhere. The answer to that was ‘no’. If you are reading this alongside the paper, note that ‘lineage’ is equivalent to ‘Sequence Cluster’ or SC. Seventy three distinct SCs were found in the complete dataset by analysis of population structure. If you don’t want to think about that too much, it’s roughly equivalent to truncating the tree at some distance and saying “genomes more closely related than this are the same SC/lineage” (only less arbitrary than that). The frequencies of SCs in the different sites were poorly correlated.
In other words, there is population structure, such that different combinations and proportions of SCs characterize each sample. What would we expect this to mean for the accessory genomes?
Well, if each SC has its own combination of accessory genes, then we would expect the frequencies of these to vary among the samples depending on which SCs were present. And given that SCs are poorly correlated among sites, the frequencies of accessory genes should be too. Right?
Wrong.
Across all compared samples, the frequencies of the accessory genes are spectacularly correlated. R2 of 0.98 or 0.99, p values microscopic. Rather than replicate the figure I’ve sketched a schematic for you (in case you can’t download the paper) I apologize in advance for my poor skills as a draughtsman.
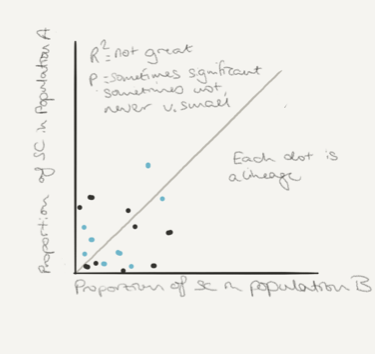
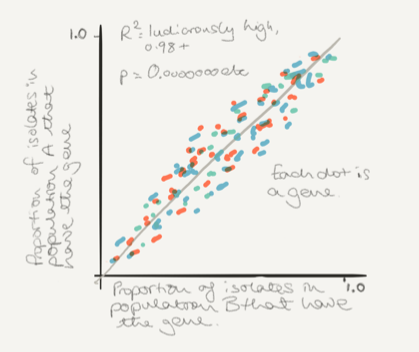
On the top, a sketch of the way the different lineages vary in frequency between populations, and on the bottom the same plot but this time for each of the accessory genes. The colors mean something in the paper, but here they are just to make it look prettier.
This is the fall off your chair moment. The point when you realize that no matter which core genomes make up the population, they only seem to be a canvas on which the accessory loci always paint the same picture.
And have you got back on your chair yet? Good, but prepare to fall off again, because the same thing happens following vaccination targeting some SCs! Pneumococcal conjugate vaccines are a fascinating intervention from an ecological point of view because they are directed at only some of the antigenic diversity in the species. Of the 90 plus serotypes that pneumococci can make and we can recognize, the most recent conjugate vaccine targets thirteen. Following vaccination the consequences for anything with a serotype in the vaccine were grave. These strains, around 50% of the population in some places, rapidly went from being “the vaccine serotypes” to being “the nearly extinct serotypes”. This was a massive selective sledgehammer to hit a population, with fully 50% of the lineages on the way out.
And yet the accessory genes don’t seem to have got the memo. Vaccine was introduced, the population took the hit, and the frequencies of accessory genes before and after vaccination were even more tightly correlated.
What makes this happen? Well the paper suggests frequency dependent selection as a possibility. These accessory genes are likely to be mobile elements, or immunogenic, and as such might experience an advantage when rare and be selected against when common. We can add to this the way the pneumococci recombine, so that the accessory loci can be shuffled into different combinations. In other words the accessory genome is indeed involved in ecological specialization, but not the way I had initially (and wrongly) thought.
This is then checked out with some modeling. I might come back to that in another blog post, but for now I will note again that although these models can explain these remarkable findings, it doesn’t mean they are the only means of explaining them. I’ve learned that lesson the hard way. In this sort of science it can be hard to tell competing explanations apart. The models do however capture some very nice things (I find elements of the stability of the population especially compelling). The results are fascinating, and among other things suggest that we might be able to predict the properties of a strain that would be successful in any given population. It would be characterized by a set of accessory loci that were presently less common than one would expect. Being able to manipulate bacterial populations effectively like this would be useful, to say the least. But we still don’t know which loci exactly are under selection, and how linkage with the core genome influences the results.
To close I want to return to my opening statement about how bacteria are different. Imagine this from the point of view of an entomologist. And instead of accessory loci let’s imagine phenotypic traits like color, or size, or any of the many things that vary in the sort of life we can see with the naked eye.
She goes for a stroll collecting samples of beetles in the woods of New England, the forests of Thailand, the lowlands of the Netherlands and an English Country Garden (quite a long stroll). Then she gets back to the lab and analyses their DNA.
Unsurprisingly she finds that those disparate locations are characterized by different species, but when she comes to investigate the way the beetles look she finds something surprising. A third of all the beetles in every sample have red wing cases, and a third yellow, while the remainder glisten a beautiful iridescent blue. When she starts to count the spots on the wings, she finds that a quarter have two spots, a third have three. Another third have no spots at all and the fraction that is left have 4 (5% if you have been keeping count). In each of the different places she visited, these were associated with different bugs. So that in the English Country Garden, the red bugs with dots were ladybirds (ladybugs for those of an American disposition) but in Thailand, there were no ladybugs. Instead the spots were found on some other beetle, and some other species was red. In Massachusetts, the ladybugs were present (according to their DNA) but in our imaginary dataset they were blue, and had no spots, while instead the dung beetles were bright yellow with spots. And all the time in each sample when you counted up these variable properties, they summed to the same proportions.
The only other thing I have to say right now is that pneumococci are only one sort of bacteria, and as I have commented before it is foolish to think that we can take them (or any other bacteria) as exemplars of all prokaryotes. Bacteria are not only different from eukaryotes; they are different from each other. We have no idea if this is a general property, or if it depends on sufficiently high recombination rates. That said, I hope this has convinced you that they are pretty weird, and wonderful.
Corander, J., Fraser, C., Gutmann, M. U., Arnold, B., Hanage, W. P., Bentley, S. D., Lipsitch, M., Croucher, N. J. (2017). Frequency-dependent selection in vaccine-associated pneumococcal population dynamics. Nature Ecology & Evolution, 1. https://doi.org/10.1038/s41559-017-0337-x